







SeeD se ha embarcado en la caracterización de la composición genética de hasta 200,000 muestras de trigo y maíz, incluyendo accesiones de bancos de germoplasma y materiales de programas de pre-mejoramiento. Este esfuerzo generará una de las bases de datos más completas que describen la diversidad genética de dos de los principales cultivos alimenticios de la humanidad. Dichos conjuntos de datos servirán como un marco unificador para los participantes del proyecto y servirán para integrar los datos fenotípicos adquiridos para diferentes subconjuntos de muestras y propósitos en diferentes lugares y tiempos.

Germinación de semillas de accesiones de maíz para extracción de ADN (30 plantas individuales por accesión; tres accesiones por bandeja. ©2012 (CIMMYT)
Las tecnologías de marcadores moleculares están en una transición de métodos de “genotipificación por ensayo”, hacia métodos de “genotipificación por secuenciación” (GBS, por sus siglas en inglés). Dicha transición es similar a la de la fotografía análoga cuando migró hacia la fotografía digital. Los métodos tradicionales de genotipificación por ensayo se basan en ensayos que detectan los polimorfismos de ADN, de manera indirecta, a través de las diferencias en la movilidad de los fragmentos de ADN en geles, la amplificación diferencial por PCR, o la hibridación diferencial a microarreglos. La caída de un orden de magnitud en el costo por base en plataformas de secuenciación de nueva generación, hace posible omitir el paso de desarrollo del ensayo y usar estas plataformas de secuenciación, no sólo para descubrir, sino también para la clasificación directa de los polimorfismos de ADN en los ensayos rutinarios de genotipificación (Elshire et al. 2001, Sansaloni et al. 2011 .

Equipo que coordina la extracción de ADN y sus aspectos logísticos. Primer fila (desde la izquierda a la derecha): Ángel Galindo Fernández, Oscar Ernesto Perez Soriano,Raymundo Blancas Márquez, Facundo Cristian Flores Cano, Ricardo Benavides Alfaro, Martin Rodriguez Espinoza; segunda fila (desde la izquierda a la derecha): María del Socorro Badillo Lopez, Noemi Ortega Jiménez, Sheila Salcido Leyva, María Fabiola Hernandez Leyva, Tania Salcido Leyva, Rosaura Almeraya Quintero, Nizayet Anahí Salas Victores, Martha Lidia Deheza Villejas; tercer fila (de izquierda a derecha): Jorge Alvarado Blancas, Irene Nayeli Benítez Hernandez, Gustavo Alberto Martinez Rodriguez, Sarah Hearne, Emmanuel Baños Galindo, Antonieta Díaz Gutiérrez. ©2012 (CIMMYT)
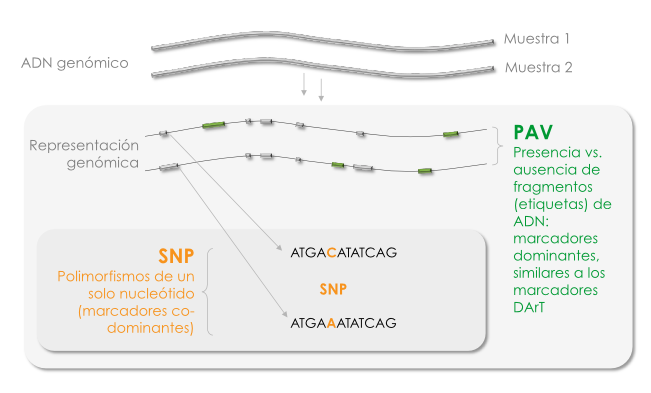
GBS se basa en re-secuenciar una pequeña y bien definida fracción del genoma (una “representación genómica”), que corresponde a 0.1-1% del genoma. Las representaciones de genomas se preparan usando métodos de “reducción de complejidad” que seleccionan de forma reproducible un subconjunto bien definido de loci de ADN (“etiquetas”) del genoma. Para SeeD, nos concentramos en métodos de reducción de complejidad, basados en enzimas de restricción (ER), métodos que son esencialmente una extensión de la tecnología de marcadores de DArT (Diversity Arrays Technology) (Jaccoud et al. 2001, Wenzl et al. 2004)
{kind=link}
Los métodos de GBS, en principio, pueden detectar dos tipos de polimorfismo de ADN:
- Polimorfismos de un solo nucleótido (SNP, por sus siglas en inglés), que se pueden identificar mediante la comparación de las diferentes variantes de una etiqueta de secuencia sencilla, amplificadas a partir del ADN genómico de diferentes individuos.
- Variantes de presencia/ausencia (PAV, por sus siglas en inglés), que son causadas por polimorfismos de ADN que conduce a la incorporación o exclusión de segmentos genómicos (etiquetas) en la representación del genoma. En el caso de los métodos de reducción de complejidad basados en ER, éstos incluyen InDels y SNP que afectan a los sitios de reconocimiento de las ER utilizadas. En caso de que se utilicen ER susceptibles a la metilación del ADN, los polimorfismos de metilación en los sitios de reconocimiento de ER también pueden causar PAV.
A pesar de algunas limitaciones, GBS tiene una variedad de ventajas estratégicas para un proyecto como SeeD. La más importante, sin duda, es la capacidad de cuantificar las relaciones o distancias genéticas con un sesgo de evaluación muchísimo menor que los microarreglos de SNP. También existe la posibilidad de estimar las frecuencias alélicas dentro de, y las distancias genéticas entre muestras mixtas de ADN, cada una derivada de una población de varios individuos genéticamente distintos (por ejemplo, muestras de variedades criollas de maíz).

Ensayo de caracterización de trigo de gran escala que también se utilizó para la extracción de ADN de una planta representativa por cada accesión. ©2012 (CIMMYT)
Dependiendo del cultivo y el propósito del ensayo de genotipificación, usamos diferentes implementaciones de GBS, desarrolladas por Diversity Arrays Technology (DArT) y el Instituto para Diversidad Genómica (IGD por sus siglas en inglés) para (a) el análisis de la diversidad del maíz (DArT), (b) el mapeo de asociación en maíz (IGD), y (c) el análisis de la diversidad y mapeo de asociación en trigo (DArT). Éstas difieren en el número de marcadores obtenidos (~ 30,000-750,000), el porcentaje de datos faltantes (~20-60%), y la posibilidad de identificar heterocigotos. Las etiquetas de secuencias analizadas por GBS pueden alinearse con una secuencia de un genoma de referencia.
En el caso del maíz, nuestros colegas de LANGEBIO están generando un genoma de referencia más versátil, volviendo a secuenciar los genomas completos de un conjunto de materiales derivados de variedades criollas tropicales. En el trigo, utilizaremos como referencia una colección de etiquetas GBS no-redundantes que se han identificado de manera reproducible en las accesiones analizadas hasta el momento. Esta colección se actualizará periódicamente y las etiquetas individuales con el tiempo se alinearán con la secuencia del genoma del trigo cuando esté disponible.
Para obtener más información, escríbanos a: seed@masagro.org
Limitaciones de GBS
- Los fragmentos (etiquetas) individuales de ADN en una representación genómica se amplifican de forma desigual, lo que tiene como resultado una distribución en “forma de L” con una larga cola de fragmentos de baja frecuencia que dan lugar a un número elevado de datos faltantes y la necesidad de imputar alelos no clasificados.
- Para el trigo hexaploide, necesitarán discriminarse los alelelos verdaderos de SNP de los homeoalelos provenientes de sub-genomas diferentes, confirmando la segregación mendeliana en poblaciones de mapeo. Hasta que se construya una base de datos suficientemente grande de SNPs verdaderos, la clasificación de alelos SNP de trigo tiene que ser conservador y excluir marcadores que pueden ser informativos. Sin embargo, estos marcadores pueden ser recuperados volviendo a analizar los datos primarios cuando se haya construido una base de datos de SNP más completa.
Ventajas estratégicas de GBS
- Siempre que las representaciones de genomas sean muestreadas a partir del genoma de una manera relativamente aleatoria, GBS esencialmente elimina el sesgo de determinación, lo que sería una limitación importante al caracterizar acervos genéticos poco explorados utilizando un conjunto fijo de marcadores. Entre las tecnologías de marcadores disponibles en la actualidad, es probable que GBS genere la imagen más precisa de la diversidad genética conservada en los bancos de germoplasma.
- Los polimorfismos de ADN son identificados y clasificados al mismo tiempo. Esta característica elimina completamente la necesidad de un costoso desarrollo de marcadores y un componente adicional de validación de marcadores en acervos genéticos secundarios y terciarios.
- GBS es escalable. La densidad de marcadores, el contenido de información por marcador (capacidad de calificar heterocigotos) y el nivel de multiplexación de las muestras pueden ser ajustados, dentro de los límites de la capacidad de secuenciación disponible con una plataforma particular.
- Los marcadores PAV evaluados por los métodos de GBS con suficiente profundidad de secuenciación son instrumentales para la cuantificación de las relaciones genéticas entre miembros de acervos genéticos distantes. Esto es porque el número de fragmentos en representaciones de genoma que se comparten y que por lo tanto pueden usarse para identificar y clasificar SNPs, disminuye a medida que la distancia genética entre los individuos aumenta.
- Las implementaciones de GBS con suficiente profundidad de secuenciación se pueden utilizar para caracterizar mezclas de ADN derivadas de poblaciones de individuos genéticamente heterogéneos. Hemos desarrollado un método para cuantificar simultáneamente (a) las distancias genéticas entre las poblaciones, y (b) las frecuencias alélicas dentro de cada una de ellas.
- GBS es un enfoque de genotipificación compatible con futuras plataformas que no “envejecerá” mucho durante el proyecto. Las secuencias pueden ser fácilmente examinadas de nuevo o integradas con datos más completos de secuencia genómicas conforme estén disponibles.
- Por último, el costo por muestra es bajo y se prevé que siga disminuyendo durante la vida útil del proyecto.